All Class II and some Class I and Class III medical devices require 510(k) approval from FDA before they are sold in the US. As such, the FDA does not provide official ‘approval’ of medical devices but clears them for sale.
How To Check if Your Medical Device Requires 510k Compliance with the FDA
If you are unsure whether your medical device requires a 510(k) submission, follow the steps below:
1. Search the FDA classification Database
The first step in determining whether your product requires a FDA 510(k) submission is to check the FDA classification database. To get started, visit: http://www.accessdata.fda.gov/scrIpts/cdrh/cfdocs/cfPCD/classification.cfm This will open a new window showing the FDA page you see below:
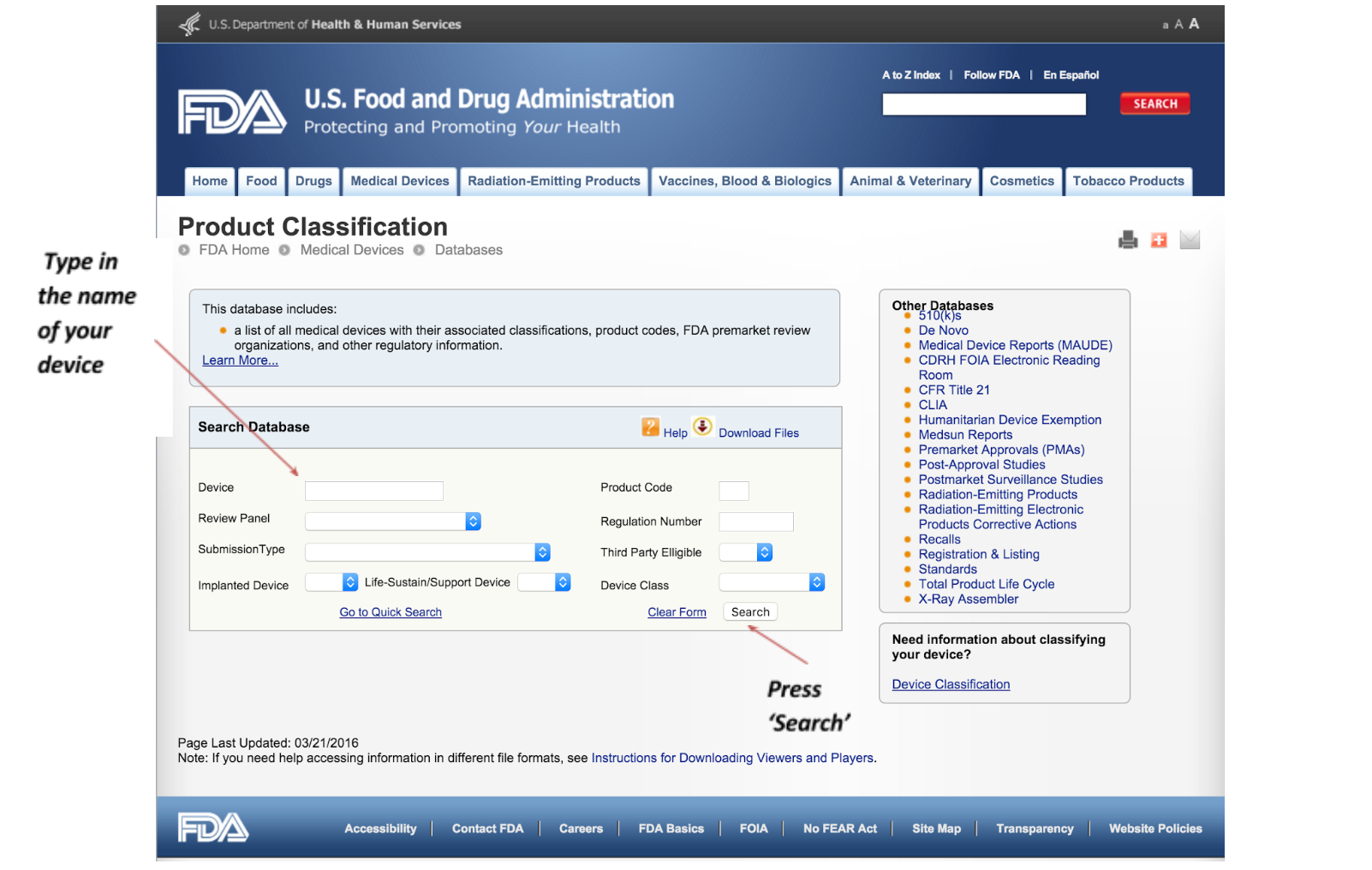
2. Review the product categories
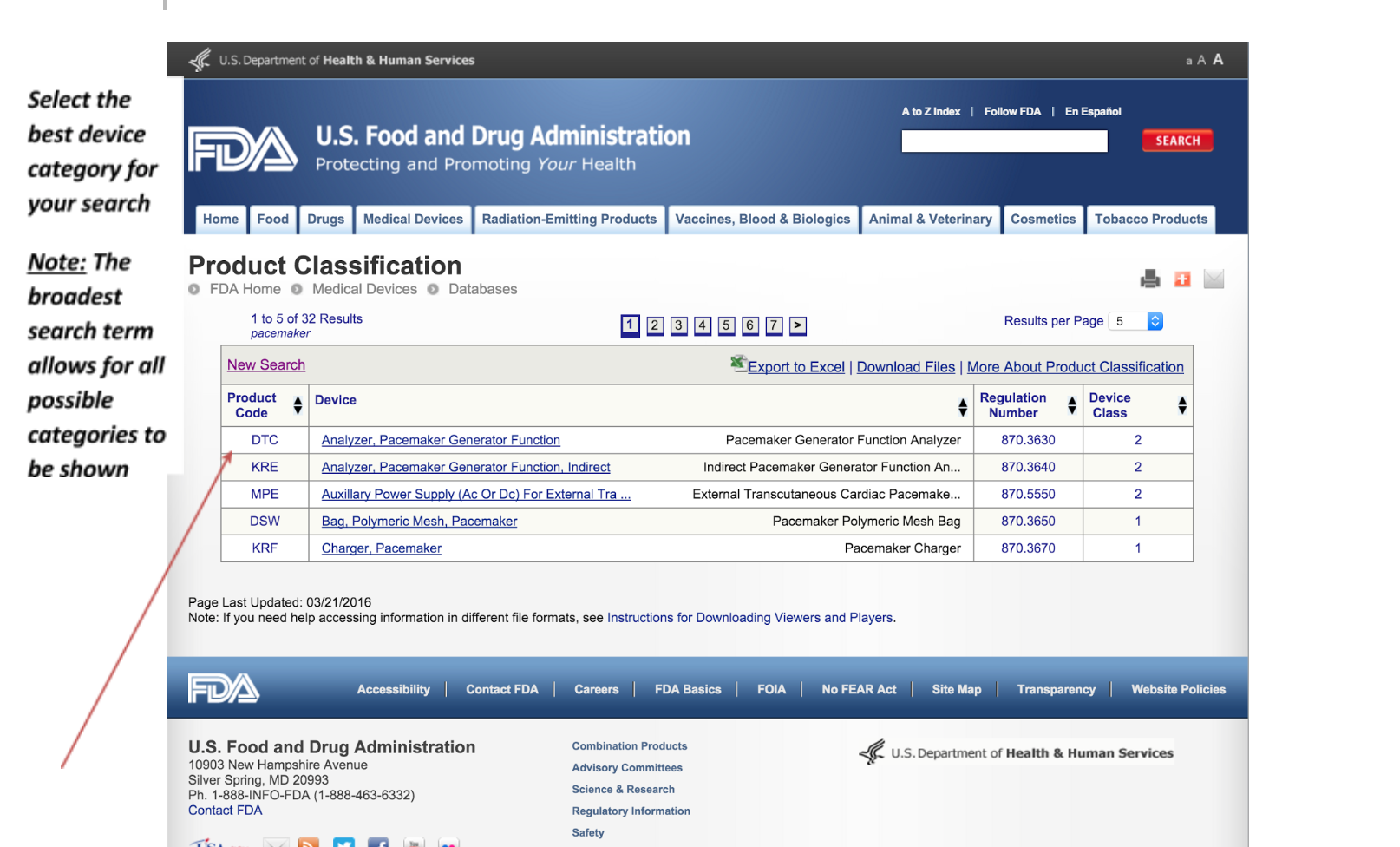
3. Review your selection

4. Read the device description

5. Go to the 510(k) database
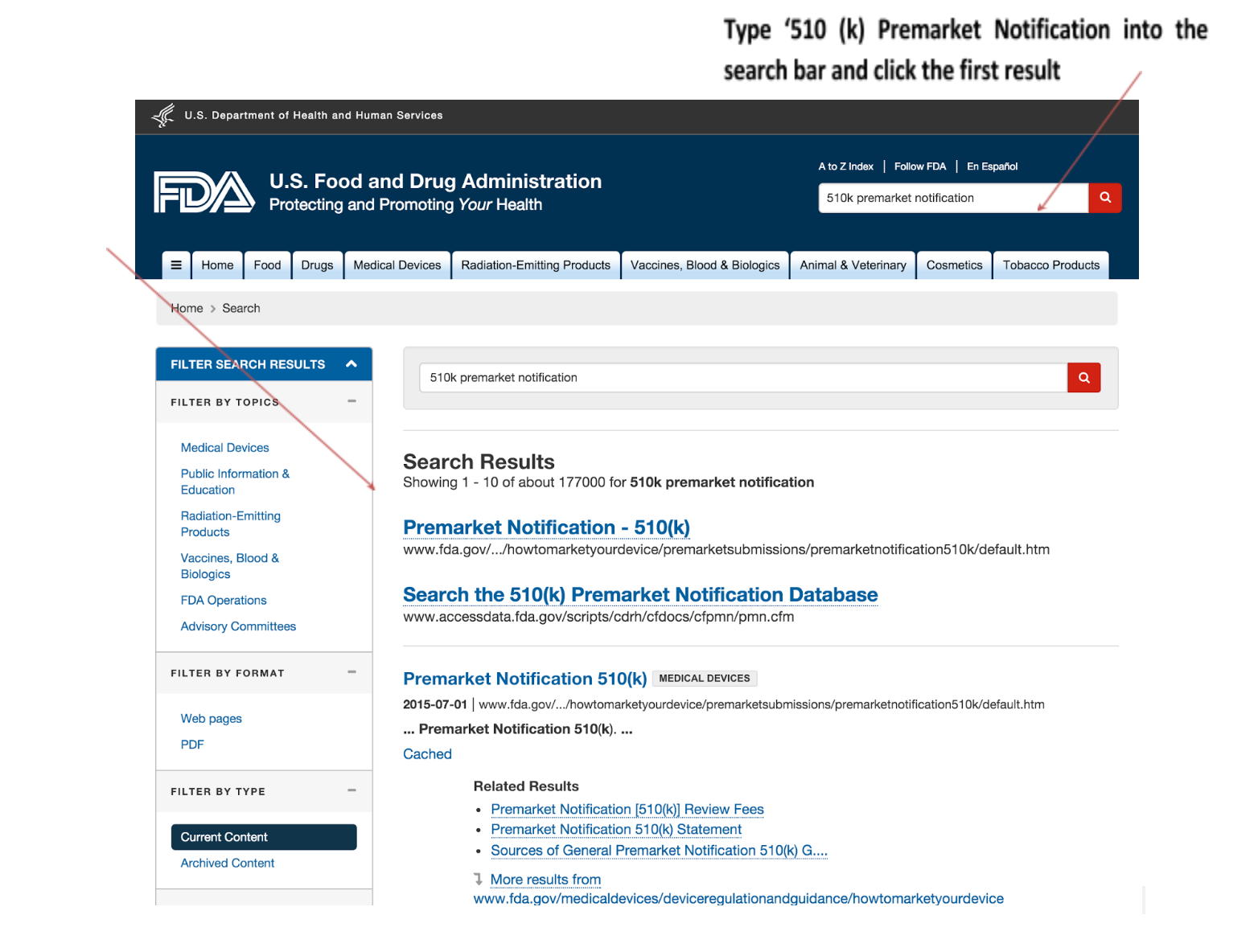
6. Review other cleared devices
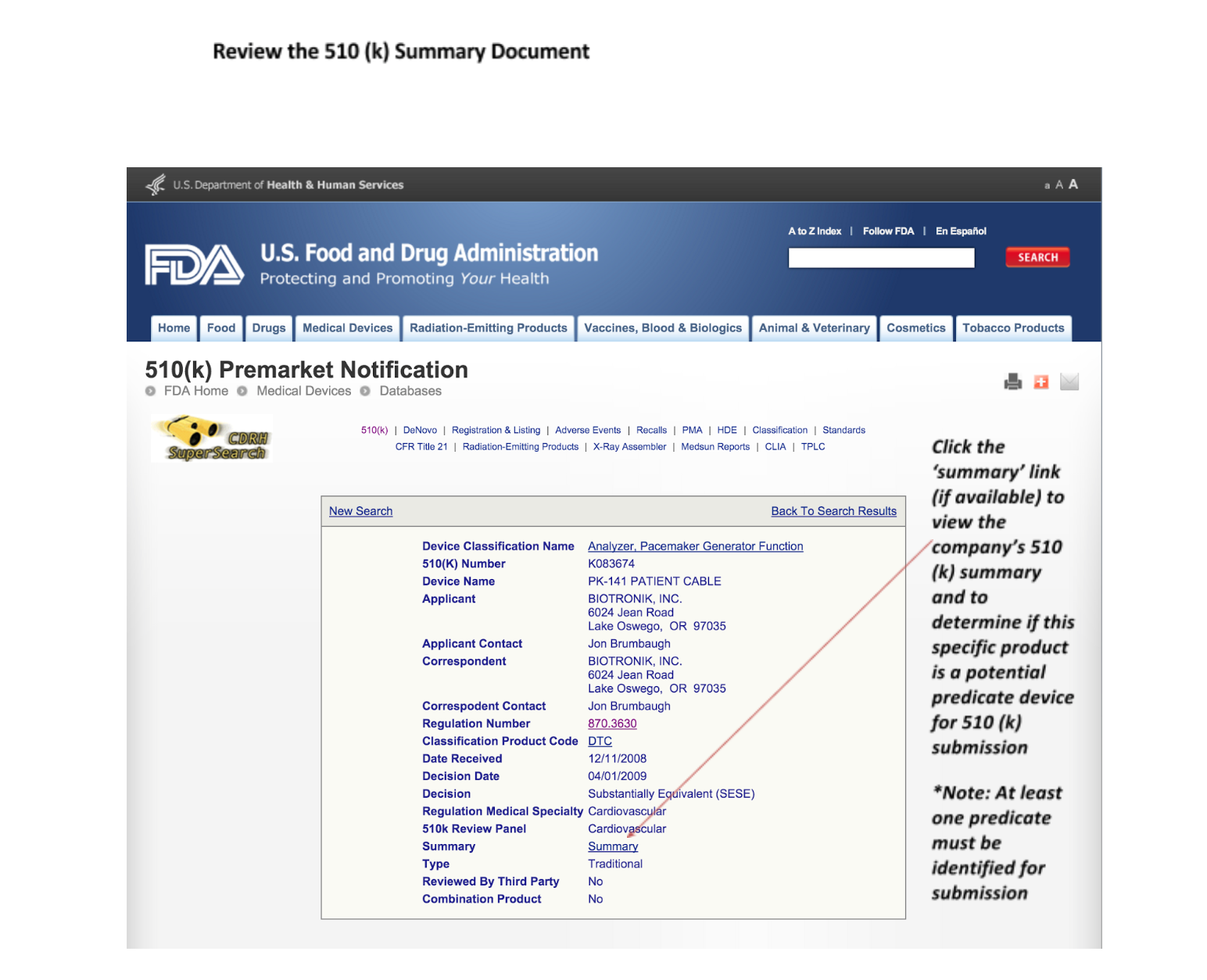
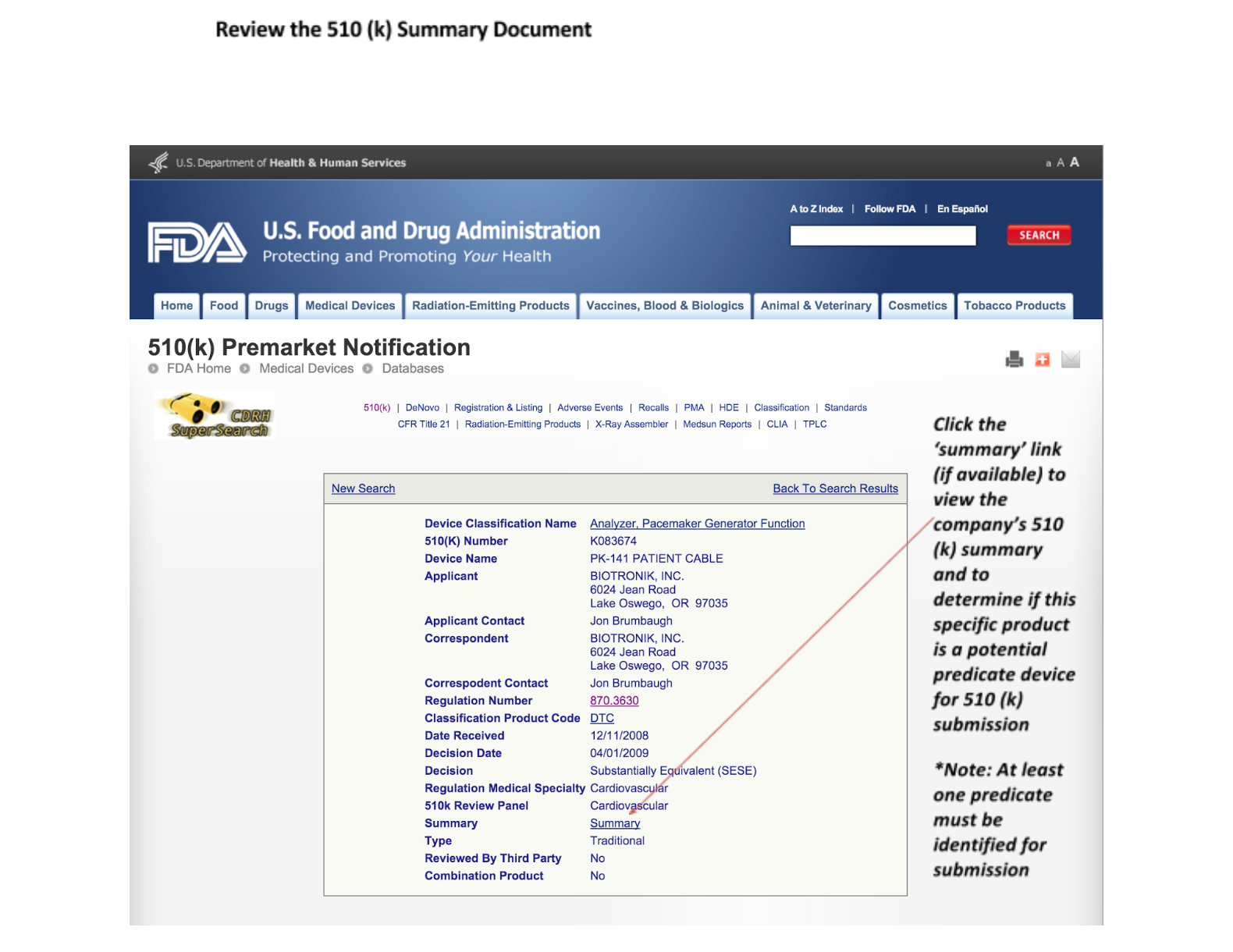
7. Read the 510(k) summary documents
If your device requires a 510(k), please follow the following steps:
CHECK FOR SIMILAR DEVICES APPROVED BY FDA
STEP 1:
Identify other medical devices similar to yours that have already received 510(k) clearance from the FDA, called “Predicate Devices.” Go to FDA Product Classification Search Database page and see the section under “Medical Devices.” Note information about each predicate device: 510(k) Number, Regulation Number and Classification Product Code.
STEP 2:
Check the Product Classification Code to find out whether any Standards and/or Guidance documents apply to your medical device. These documents are additional requirements you must provide to complete the 510(k) process.
SUBMISSION
STEP 3:
After identifying the predicate devices and gathering information about them, prepare and submit the 510(k) to the FDA. Please note that the FDA charges a fee to review your submission.
REVIEW & CLEARANCE
STEP 4:
FDA will review your submission within 90 days. You may be required to submit additional information during this period. FDA stops the ‘clock’ during this time and resumes when you have provided all answers to their queries. Please note that ‘review’ does not mean ‘clearance’.
If your device is cleared, you will receive a 510(k) clearance letter by post from the FDA with an assigned 510(k) number. The letter states that FDA “have determined that your device is substantially equivalent to legally marketed predicate devices – and you may therefore begin to market your device subject to the general controls provisions of the Food, Drug and Cosmetics Act.”
This letter does not mean that FDA ‘approves’ your device. The FDA is just confirming that your medical device is considerably similar to the predicate device(s) selected in your 510(k), and which has already been cleared for sale by the FDA, and that you are now cleared to sell your medical device. FDA will not provide a certificate, but this letter will be available on the FDA database as proof to your customers that your product is cleared for sale in the US.
REGISTRATION
STEP 5:
After receiving your FDA 510(k) ‘clearance’ letter, visit the FDA Device Registration and Listing page and register your device and company with the FDA using your assigned 510(k) number. If you are located outside the US, you must appoint a US Agent at this time.
FDA COMPLIANCE
STEP 6:
Now that your device has been cleared and you have received an assigned FDA 510(k) number for your device which does not expire, you are required to be compliant with all FDA regulations while you are selling the device in the US. The FDA may visit for a facility inspection at any time to check your compliance with the Quality Systems Regulation (QSR), 21 CFR Part 820.
